A R/MATLAB package to construct and compare single-cell gene regulatory networks (scGRNs) using single-cell RNA-seq (scRNA-seq) data sets collected from different conditions based on machine learning methods. scTenifoldNet uses principal component regression, tensor decomposition, and manifold alignment, to accurately identify even subtly shifted gene expression programs.
scTenifoldNet requires numpy and scipy from the Python programming language, to install them, we recommend to install miniconda Python: https://docs.conda.io/en/latest/miniconda.html.
After install the Phyton dependencies, install scTenifoldNet, using the following command:
library(devtools)
install_github('cailab-tamu/scTenifoldNet')
library(scTenifoldNet)
| Code | Function |
|---|---|
| installPyDependencies | Install the Phyton dependencies using the reticulate package |
| scQC | Performs single-cell data quality control |
| cpmNormalization | Performs counts per million (CPM) data normalization |
| pcNet | Computes a gene regulatory network based on principal component regression |
| makeNetworks | Computes gene regulatory networks for subsamples of cells based on principal component regression |
| tensorDecomposition | Performs CANDECOMP/PARAFAC (CP) Tensor Decomposition |
| manifoldAlignment | Performs non-linear manifold alignment of two gene regulatory networks |
| dCoexpression | Evaluates gene differential coexpression based on manifold alignment distances |
| scTenifoldNet | Gonstruct and compare single-cell gene regulatory networks (scGRNs) using single-cell RNA-seq (scRNA-seq) data sets collected from different conditions based on principal component regression, tensor decomposition, and manifold alignment. |
Once installed, scTenifoldNet can be loaded typing:
library(scTenifoldNet)
Here we simulate a dataset of 2000 cells (columns) and 100 genes (rows) following the negative binomial distribution with high sparsity (~67%). We label the last ten genes as mitochondrial genes ('mt-') to perform single-cell quality control.
nCells = 2000
nGenes = 100
set.seed(1)
X <- rnbinom(n = nGenes * nCells, size = 20, prob = 0.98)
X <- round(X)
X <- matrix(X, ncol = nCells)
rownames(X) <- c(paste0('ng', 1:90), paste0('mt-', 1:10))
We generate a perturbed network modifying the expression of genes 10, 2, and 3 and replacing them with the expression of genes 50, 11, and 5.
Y <- X
Y[10,] <- Y[50,]
Y[2,] <- Y[11,]
Y[3,] <- Y[5,]
Here we run scTenifoldNet under the H0 (there is no change in the coexpression profiles) using the same matrix as input and under the HA (there is a change in the coexpression profiles of some genes) using the control and the perturbed network.
outputH0 <- scTenifoldNet(X = X, Y = X,
nc_nNet = 10, nc_nCells = 500,
td_K = 3, qc_minLibSize = 30,
dc_minDist = 0)
outputHA <- scTenifoldNet(X = X, Y = Y,
nc_nNet = 10, nc_nCells = 500,
td_K = 3, qc_minLibSize = 30,
dc_minDist = 0)
As is shown below, under the H0, none of the genes shown a significative difference in coexpression profiles using an FDR cut-off of 0.1, but under the HA, the 6 genes involved in the perturbation (50, 11, 2, 10, 5, and 3) are identified as perturbed.
head(outputH0$diffCoexpression, n = 10)
# gene distance Z p.value p.adj
# 23 ng23 3.070131e-15 2.321093 0.01014092 0.5925054
# 2 ng2 3.028738e-15 2.261954 0.01185011 0.5925054
# 19 ng19 2.912207e-15 2.093459 0.01815409 0.6051362
# 34 ng34 2.769999e-15 1.883624 0.02980793 0.7227582
# 9 ng9 2.617076e-15 1.652429 0.04922353 0.7227582
# 79 ng79 2.602655e-15 1.630314 0.05151755 0.7227582
# 61 ng61 2.582711e-15 1.599635 0.05483975 0.7227582
# 71 ng71 2.489774e-15 1.455239 0.07280162 0.7227582
# 20 ng20 2.479350e-15 1.438890 0.07509082 0.7227582
# 16 ng16 2.461000e-15 1.410037 0.07926437 0.7227582
head(outputHA$diffCoexpression, n = 10)
# gene distance Z p.value p.adj
# 50 ng50 0.03122694 3.016417 0.001278908 0.05348371
# 11 ng11 0.03027085 2.955685 0.001559876 0.05348371
# 2 ng2 0.03013548 2.946972 0.001604511 0.05348371
# 10 ng10 0.02759308 2.777666 0.002737543 0.06065138
# 5 ng5 0.02657637 2.706716 0.003397617 0.06065138
# 3 ng3 0.02625458 2.683841 0.003639083 0.06065138
# 31 ng31 0.01309219 1.490124 0.068095790 0.92871127
# 96 mt-6 0.01099588 1.223016 0.110661811 0.92871127
# 6 ng6 0.01067100 1.178311 0.119336356 0.92871127
# 59 ng59 0.01063989 1.173978 0.120201927 0.92871127
Results can be easily displayed using quantile-quantile plots. Here we labeled in red the identified perturbed genes with FDR < 0.1.
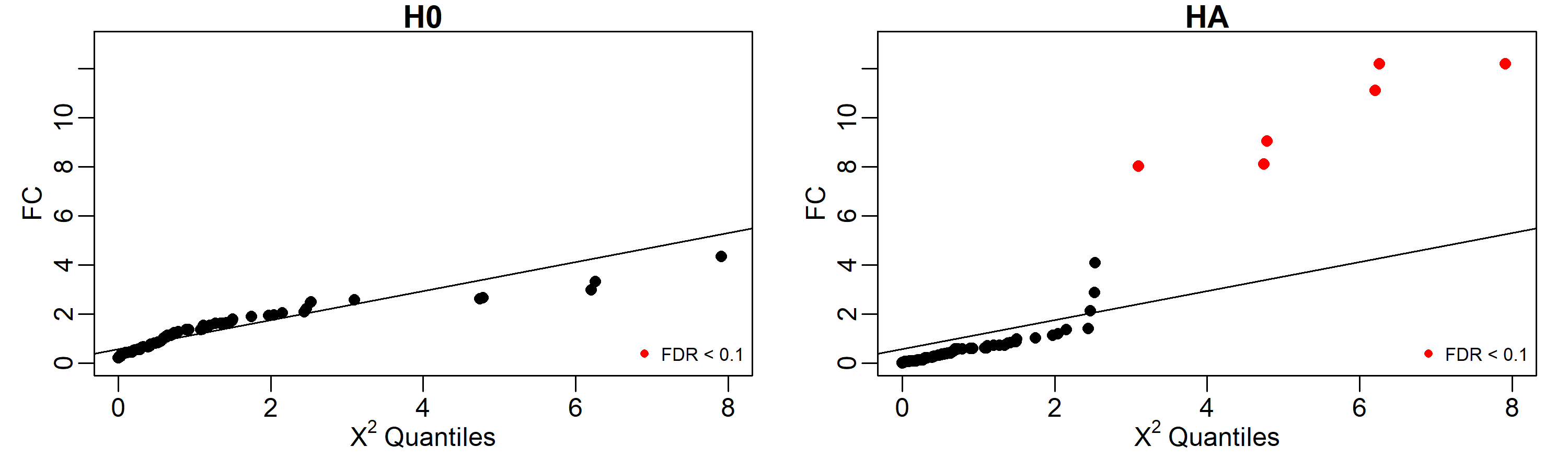
par(mfrow=c(1,2), mar=c(3,3,1,1), mgp=c(1.5,0.5,0))
geneColor <- ifelse(outputH0$diffCoexpression$p.adj < 0.1, 'red', 'black')
qqnorm(outputH0$diffCoexpression$Z, pch = 16, main = 'H0', col = geneColor)
qqline(outputH0$diffCoexpression$Z)
legend('bottomright', legend = c('FDR < 0.1'), pch = 16, col = 'red', bty='n', cex = 0.7)
geneColor <- ifelse(outputHA$diffCoexpression$p.adj < 0.1, 'red', 'black')
qqnorm(outputHA$diffCoexpression$Z, pch = 16, main = 'HA', col = geneColor)
qqline(outputHA$diffCoexpression$Z)
legend('bottomright', legend = c('FDR < 0.1'), pch = 16, col = 'red', bty='n', cex = 0.7)