Let's benchmark quantum chemistry packages! Who will be the winner?
Gaussian 09, Gaussian 16, ORCA 4, Turbomole 7, NWChem 7, Q-Chem 5, GAMESS-US 2020, PySCF 1.7, Psi4 1.3, Firefly 8, Dalton 2018
No.1 : General test
- Molecule: C20
- Properties: 20 atoms, charge = 0 and spin mult. = 1
- Task: Single-point energy calculation
- Method: DFT B3LYP/6-31G(d)
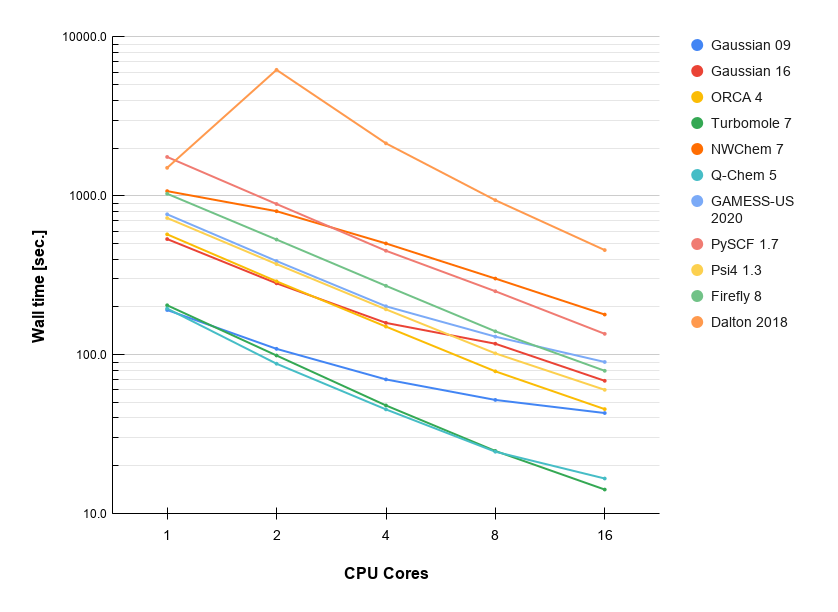
| No. cores | Gaussian 09 | Gaussian 16 | ORCA 4 | Turbomole 7 | NWChem 7 | Q-Chem 5 | GAMESS-US 2020 | PySCF 1.7 | Psi4 1.3 | Firefly 8 | Dalton 2018 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 191.4 | 535.1 | 573.0 | 205.0 | 1071.9 | 195.6 | 766.2 | 1758.9 | 726.2 | 1031.8 | 1502.0 |
| 2 | 109.0 | 281.8 | 290.0 | 99.0 | 801.2 | 87.9 | 389.1 | 889.3 | 372.2 | 531.2 | 6215.0 |
| 4 | 70.0 | 158.8 | 150.5 | 48.0 | 502.2 | 45.3 | 202.0 | 451.8 | 193.2 | 271.8 | 2142.0 |
| 8 | 52.0 | 117.4 | 78.7 | 24.8 | 301.9 | 24.6 | 130.1 | 251.2 | 102.1 | 140.5 | 940.0 |
| 16 | 43.0 | 68.6 | 45.5 | 14.2 | 179.2 | 16.7 | 90.1 | 135.5 | 60.2 | 79.3 | 456.0 |
No.2 : SCF energy cutoff 1e-8
- Molecule: C20
- Properties: 20 atoms, charge = 0 and spin mult. = 1
- Task: Single-point energy calculation
- Method: DFT B3LYP/6-31G(d)
- SCF cutoff: 1e-8
The result is coming ...
No.3 : Gaussian 16 with GPU acceleration
- Molecule: C20 (xyz coordinate)
- Properties: 20 atoms, charge = 0 and spin mult. = 1
- Method: DFT B3LYP/6-31G(d)
- 4 x NVIDIA Tesla V100 SXM2 32 GB / Intel Xeon Gold 6132 CPU @ 2.60GHz
| No. actual used CPU cores | No GPU | 1 GPU | 2 GPUs | 4 GPUs |
|---|---|---|---|---|
| 1 | 757.2 | 181.4 | 153.9 | 107.2 |
| 2 | 406.5 | 157.5 | 134.2 | 97.5 |
| 4 | 241.2 | 146.5 | 123.3 | 61.9 |
| 8 | 164.6 | 88.4 | 70.8 | 56 |
| 16 | 88.9 | 63.7 | 57.1 | 47.4 |
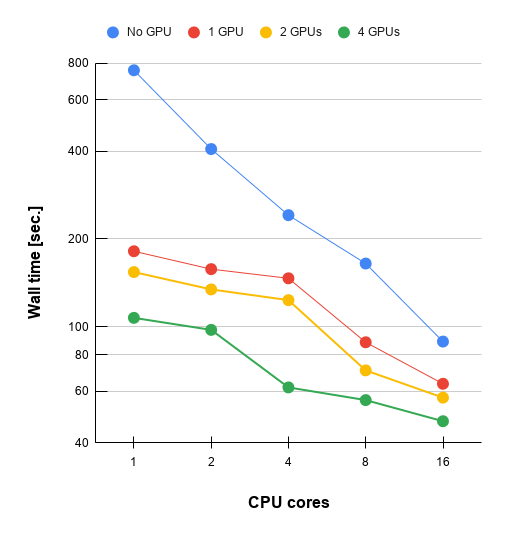
Comment: Note that one CPU will be sacrified to control one GPU. It means that CPUs used as GPU controllers do not participate as compute nodes during the parts of the calculation that are GPU-parallel. For example, for running calculation on 16 CPUs and 4 GPUs, the total number of CPUs that used for this job is 20 CPUs.
No.4 : MP2 test
- Molecule: Aspirin (xyz coordinate)
- Properties: 21 atoms, charge = 0 and spin mult. = 1
- Task: Single-point energy calculation
- Method: MP2/cc-pVTZ
The result is coming ...
No.5 : CCSD(T) test
- Molecule: Cyclopropane (xyz coordinate)
- Properties: 9 atoms, charge = 0 and spin mult. = 1
- Task: Single-point energy calculation
- Method: CCSD(T)/cc-pVTZ
The result is coming ...
- Default DIIS
- Using default values of each package for convergence criteria, integral grids, etc. Unless otherwise stated.
- In ORCA, Psi4, and Turbomole, RI (or DF) approaximation is turned off
- GAMESS and Firefly use the SCF density change as a convergence criteria. For more details please check CONV keyword.
- Details of program and packages compilations: click here
- Specification of compute node: click here
Changes in Defaults between Gaussian 09 and Gaussian 16:
a. Integral accuracy is 10^-12 rather than 10^-10 in G09
b. The default DFT grid for general use is UltraFine rather than
FineGrid in G09; the default grid for CPHF is SG1 rather than
CoarseGrid.
c. SCRF defaults to the symmetric form of IEFPCM (not present in G09)
rather than the non-symmetric version.
d. Physical constants use the 2010 values rather than the 2006 values
in G09.
We do not aim at showing the preference of one package over another, users should choose packages according to their:
- Level of familiarity
- Type of calculation
- Hardwares availability
which are more important than a simple speed comparison.
- DFT - SCF with the energy convergence cutoff of 1e-8
- MP2/cc-pVTZ calculation
- CCSD(T)/cc-pVTZ calculation
- Performance benchmarking on the robustness of geometry optimization engine
- Gradient calculation
All suggestions, comments, pull requests, etc are welcome. Please write us at r2compchem@gmail.com.